Cross-contamination is a potential risk within the vaccine manufacturing process, especially if different products are processed in the same controlled rooms. Cross-contamination can occur if required controls are not in place to eliminate risks that could impact product quality and, most of all, compromise human lives.
Miss the first post in the series? Read "Sources of Cross-Contamination in Vaccine Production"
With the unexpected novel COVID-19 outbreak, the vaccine industry direction shifted dramatically and is now facing challenges due to high demand in vaccine volume manufacturing. This prompts concern about contamination controls for the biotech and vaccine production facilities. Under the circumstances, it may be appropriate to rethink some existing contamination control programs both for risks to the product and to the individuals working in these areas.
Where Control of Contamination Stands Today
Contamination is likely to be most significant in products administered by injection or applied to open wounds or those given in large doses and/or over a long time. With the appearance of COVID-19, contract manufacturing operations (CMOs) are currently facing tremendous pressure and tight timelines to prepare modular clean suites and associated process controls to launch the large-scale production of vaccines for commercial use. This increases potential risks in vaccine production if robust quality systems, discipline process controls, and periodic monitoring programs are not in place.
The cell gene therapy industry was also hit by the pandemic, and this industry is now merging efforts with the vaccine industry to support the high demand. Hospital units were overwhelmed with COVID-19 cases, but this combined effort will not only allow hospitals to resume cell gene therapies without interruption but will also support the large-scale vaccine production process, which needs 9 billion vaccines by the end of 2021.
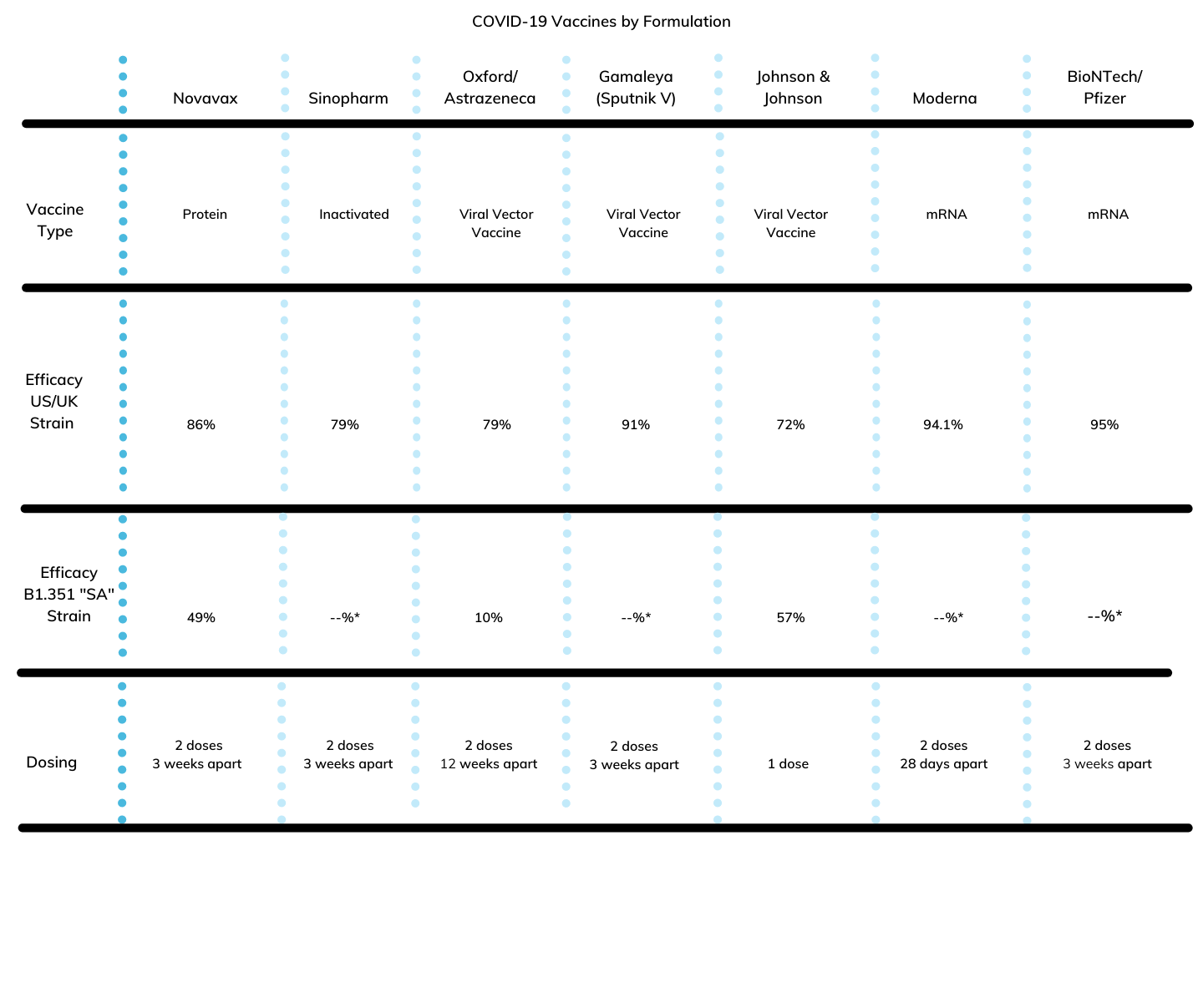 Figure 1 – Table COVID-19 vaccines being produced, along with their type, efficacy across strains, dosing, and storage requirements.
Figure 1 – Table COVID-19 vaccines being produced, along with their type, efficacy across strains, dosing, and storage requirements.
* B1.351 “SA” Strain – Another variant of SARS-CoV-2 found in South Africa (known as 20H/501Y.V2 or B.1.351) emerged independently of B.1.1.7. Not Reported.
Fortunately, with updated technologies, process controls, and other quality risk management tools, contamination and cross-contamination can be prevented through the viral controls discussed below.
Viral Controls & the Prevention of Cross-Contamination
Viral Controls
A viral contamination can shut down a biopharmaceutical plant for months. This impacts manufacturing operations, causing significant business disruption, and ultimately threatening drug supply and patient safety. Due to the high stakes, there are two main phases in the vaccine production process: upstream and downstream.
The upstream phase has the highest risk since any contamination can be carried over to the next phase. Most manufacturers focus on incorporating viral-reduction steps into their downstream processes; however, including key viral clearance steps into an upstream process can reduce the risk of viral contamination significantly. Understanding the risk of viral contamination from individual components in the upstream process can be challenging, but this risk must be considered in the context of both the likelihood of contamination, and the probability that contamination will be detected.
Adventitious viruses can be introduced into a manufacturing process through handling of cell cultures or media and the use of contaminated biologics or nonbiologic reagents. For example, some of the influenza and the novel COVID-19 vaccines are manufactured using specific real virus gene sections that are inactivated and encapsulated into the same viral capsule or vector. Viral vectors are utilized as a vehicle to be introduced via injection to patients once the solution is filtered and purified during the downstream process.
As for the gene-cell industry, the same process happens when genes (for example, editing, correction, inactivation) and cells (for example, Cart-T Cells, replacement, destruction) are manipulated to obtain the final formulation and filling of IV bags to be administered to patients during the therapies. For the vaccine and gene-cell industries, the upstream and downstream phases are similar. The upstream processes include cell line development, cell culture, and viral harvest. The downstream processes include purification, formulation, and filling.
A Prevention Strategy
On a high level, there is a holistic approach to viral safety, and prevention relies on a multilayered strategy involving three principles:
- Prevent: Careful selection and pre-treatment of raw materials to prevent viruses from entering upstream processes
- Detect: Testing for the presence of viruses
- Remove: Implementing appropriate purification and filtration technologies to remove viruses downstream
Upstream steps may include UV-C inactivation and HTST (High Temperature Short Time). Exposure to low pH or solvent/detergent treatment steps deactivates enveloped viruses downstream. Downstream steps typically include virus-removing (nano-) filters designed for the removal of almost all virus types through a size-exclusion mechanism and chromatographic resins in column or membrane configurations.
The introduction of single-use and automation technologies can also safely accelerate vaccine development and production. These are used most often in the operations of CMOs and Contract Development Manufacturing Operations (CDMOs). For example, stirred-tank bioreactors represent a key technology because it potentially reduces cross-contamination by reducing cleaning cycles. These are optimal tools for each step in upstream biology, enabling parallel control of several bioreactors for efficient and reproducible optimization of different process parameters. Vaccine product quality has benefitted from applying programs with automated responses, such as feeding cycles and pH control adjustments for the vaccine and cell gene/viral vector therapies in the CMO/CDMO industries. Our EGLS Subject Matter Expert (SME) Process Engineer Consultants have been successfully applying these process improvements programs at most top CMO/CDMO sites.
Further Steps to Prevent Cross-Contamination in the Vaccine Industry
Besides viral controls, contamination is the unwanted introduction of impurities into starting material, intermediates, or Active Product Ingredient (API). Products or substances are tainted by other drug substances or products. This can occur during storage, transport, production, sampling, or packaging.
“Prevention of contamination and cross-contamination involves requires process developers to identify sources of potential risks and to define the required control and actions to reduce and/or eliminate them.”
As shared in my previous post, the main sources of cross-contamination in drug manufacturing are human beings, air, equipment, water, and raw materials. Human beings carry normal flora such as Staphylococcus aureus and may serve as the main source of microbial cross-contamination during the manufacturing process. Therefore, personnel flow, equipment flow, and the testing process (including lab testing and/or incoming material testing) require a rigorous plan that follows Good Manufacturing Practices (GMP) to avoid starting any cross-contamination that may impact the next steps in the process.
Prevention of contamination and cross-contamination requires process developers to identify sources of potential risks and to define the required control and actions to reduce and/or eliminate them. Some functional elements guiding cross-contamination quality risk assessment during the biopharma process are:
- Quality Risk Management (QRM): This provides different risk management tools such as Failure Mode Effect Analysis (FMEA) and Hazard Analysis Critical Control Point (HACCP), etc. Cross-contamination risk assessment will cover the four pathways of cross-contamination: Mix-up, Retention, Airborne, and Mechanical Transfer. An approved QRM will be also useful for any new product to be introduced to the facilities.
- API Handling: PPE and containment requirements, defining facility and equipment resources.
- Cleanability Assessment: Validation of the cleaning method based on defined cleaning limits and equipment assessment.
- Cleaning Validation Cycle: A matrix including a list of dedicated or single-use disposable products and cleaning procedures.
- Containment Plan: Specific material flow procedures, isolators, dedicated equipment, facilities, suites, weighing, enclosure integrity, inspection, etc.
- Training Program: Conducting regular training sessions, including a “lessons learned” session from previous excursions.
Facilities design is also critical for preventing cross-contamination in the sterile/aseptic environment as well. Air carries a lot of organic and inorganic materials, which may be potential contaminants in pharmaceutical manufacturing. To avoid air contamination during manufacturing, every site must provide appropriately designed airlocks, pressure differentials, and air supply extraction systems, which also apply to modular clean suites. Pipes used for conveying distilled or de-ionized water and, where appropriate, other water pipes should be sanitized and stored according to written procedures that detail the action limits for microbiological contamination and the measures to be taken.
New technologies that are being developed can also help mitigate risks of contamination and cross-contamination. CMO/CDMO biotech companies are using modern practices like single-use and sterile connectors, which minimize human interventions throughout the manufacturing processes.
Several other technical and organizational elements can help you avoid cross-contamination:
- A dedicated manufacturing facility (premises and equipment).
- Self-contained production areas having separate processing equipment and separate heating, ventilation, and air-conditioning (HVAC) systems. It may also be desirable to isolate certain utilities from those used in other areas.
- Design of manufacturing processes, premises, and equipment to minimize opportunities for cross-contamination during processing, maintenance, and cleaning.
- Dedicating the whole manufacturing facility or a self-contained production area on a campaign basis (dedicated by separation in time) followed by a cleaning process of validated effectiveness.
- Keeping specific protective clothing inside areas where products with a high risk of cross-contamination are processed.
- Cleaning verification after each product campaign should be considered as a detectability tool to support effectiveness of the Quality Risk Management approach for products deemed to present higher risk.
- Depending on the contamination risk, verification of cleaning of non-product contact surfaces and monitoring of air within the manufacturing area and/or adjoining areas. This demonstrates effectiveness of control measures against airborne contamination or contamination by mechanical transfer.
- Specific measures for waste handling, contaminated rinsing water, and soiled gowning.
- Recording of spills, accidental events, or deviations from procedures.
- Design of cleaning processes for premises and equipment so that the cleaning processes in themselves do not present a cross-contamination risk.
- Design of detailed records for cleaning processes to ensure completion of cleaning in accordance with approved procedures and use of cleaning status labels on equipment and manufacturing areas.
- Use of common general wash areas on a campaign basis.
- Supervision of working behavior to ensure training effectiveness and compliance with the relevant procedural controls.
Summary
As of this post, no infectious virus has been transmitted to a patient by a biopharmaceutical derived from a cell line or other source of contamination. Occasionally, however, viral contamination has been detected in process intermediates (e.g., bulk harvests). Such contaminants were detected long before products reached patients, so no virus transmission to a patient occurred.
A manufacturing process designed with a viral safety program will include specific robust virus inactivation and removal procedures, as well as purification unit operations that provide further viral reduction. The European Medicines Agency (EMA), the U.S. Food and Drug Administration (FDA), and other authorities include guidelines focused on testing and evaluating the viral safety of biotechnology products derived from characterized cell lines of human or animal origin. Guidelines provide a general framework for testing production cell banks and bulk harvests for viral contaminants and designing viral clearance evaluation studies.
To avoid contaminated vaccines, it is essential to demonstrate that equipment is free of potential contaminants that could impact the quality of end-products. A risk-based approach program, along with SME contribution to new technologies and approaches, is also crucial to consistently providing product safety and quality. Finally, measures to prevent cross-contamination and their effectiveness should be reviewed periodically according to set procedures.
CMO/CDMO operational facilities offer a great advantage with the implementation of clean suites and associated controls for the vaccine and cell gene (monoclonal) therapy industries. With COVID-19, these industries are ramping up fast due to the need of commercial vaccines and patients needing therapies/treatments for specific compromising diseases. Monoclonal treatment is bringing a new breakthrough in the biotech industry as a new way of treatment for COVID-19. Read my follow-up blog post, "Cell Gene Therapy and Viral Vectors," to find out more about cell gene and viral vector applications for the treatment of compromising disease and how it could be effective for COVID-19.
Having EG Life Sciences in your corner can help you implement best practices that can improve your vaccine production processes without compromising safety.
See how EG Life Sciences can help!

Image Sources
- https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html
- https://www.nytimes.com/live/2021/02/07/world/covid-19-coronavirus
- https://www.nytimes.com/2021/01/29/health/covid-vaccine-johnson-and-johnson-variants.html
Other Sources
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/non-penicillin-beta-lactam-drugs-cgmp-framework-preventing-cross-contamination. Accessed 29 March 2021.
- https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-virus-safety-evaluation-biotechnological-investigational-medicinal-products_en.pdf. Accessed 29 March 2021.
- https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-4/chapter_3.pdf. Accessed 29 March 2021.
- https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-emerging-variants.html. Accessed 1 April 2021.
- Variant Chart SARS-CoV2 from CDC (.xlsx). Accessed 1 April 2021.
